助力罕见病药物研发:国际罕见病研究联合会制定《孤儿药开发指南》
转自冰桶挑战基金会公众号:china_icf
编者按:国际罕见病研究联合会(International Rare Disease Research Consortium, IRDiRC)由多个国家机构、国际组织、非盈利基金会、公司(包括制药公司及生物技术公司)、患者组织、科研机构等组成,创立于2011年,致力于推动全球范围内的罕见病研究合作。该机构在2017年制定了十年目标,其中之一即是在2017-2027年的十年间促进1000种新的罕见病疗法问世。最近,IRDiRC制定了一本指南,通过将现有工具组织成标准化框架,促进罕见疾病的药物开发,现编译如下:
助力罕见病药物研发:IRDiRC制定孤儿药开发指南
作者:Anneliene Hechtelt Jonker, Virginie Hivert, Michela Gabaldo, Liliana Batista, Daniel O’Connor, Annemieke Aartsma-Rus, Simon Day, Ken Sakushima & Diego Ardigo
编译:Richard
全世界有数百万人受到罕见疾病的影响,但据估计只有不到5%的已知罕见疾病有至少一种经批准的治疗药物[1]。随着各国监管和经济激励措施的逐步普及,针对罕见疾病的药物研发项目数量正在稳步上升[2]。国际罕见疾病研究联合会(International Rare Disease Research Consortium, IRDiRC)早先曾制定计划,预期到2027年为1000种新的罕见疾病找到对应疗法[3]。但以目前的药物开发速度,该目标难以实现。
罕见病药物的开发存在着诸多挑战。我们对大多数疾病的认识仍非常有限,罕见病所对应的小患者群难以获得足够效力的疗效和安全性数据,药物研发公司以及医保支付机构都需要在罕见病药物方面应对特殊的财务可持续性的风险等。以上这些原因,造成目前传统的药物开发模式在罕见病领域明显效率低下,亟待一个新的研发框架以加速罕见病药物的研发过程。
在过去的十年中,各类罕见病支持组织综合采用了包括科学研究、政策激励、监管举措、社会资源等多种途径以加快罕见疾病的药物开发,典型措施包括利益攸关方沟通方案、创新的临床试验方案和执行策略,以及新的整体药物开发策略。然而,这些行之有效的工具尚未被所有罕见病支持组织系统和持续地使用。随着新一代利益攸关方,包括慈善机构和病人组织的出现,这一问题日益突出。这些利益攸关方往往对未满足临床需求有着的强烈的关注和深刻的理解,但在药物开发的复杂性方面经验有限。
罕见病药物开发的IRDiRC工具箱
为了改善这一状况,IRDiRC的疗法科学委员会(Therapies Scientific Committee)启动了《孤儿药开发指南》(Orphan Drug Development Guidebook, ODDG)项目。该项目旨在充分地介绍美国、欧洲和日本现有的罕见病药物开发工具,形成系统化的文档框架,从而构成能够指导罕见病药物开发的多个"模块(Building Blocks, BB)"。ODDG项目成立了一个由24位利益攸关方代表、罕见病药物开发专家组成的工作组。该工作组中囊括了学术界、监管机构、制药工业、医务工作者、患者、公共/非营利研究资助者和咨询公司(图1)。
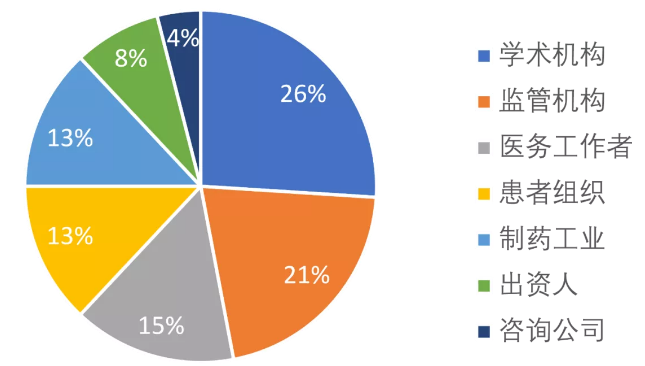
图1
IRDiRC ODDG工作组成员:Annemieke Aartsma-Rus (Leiden 大学医学中心), Alessandro Aiuti (Ospedale San Raffaele, 意大利), Diego Ardigo (Chiesi Farmaceutici S.p.A.), Dimitrios Athanasiou (世界DMD组织, 希腊), Laurie Conklin (RevaraGen, 美国), Seng Cheng (辉瑞制药公司, 美国), Robin Conwit (美国国立卫生研究院 神经疾病与中风研究中心), Simon Day (Clinical Trials Consulting and Training Limited, 英国), Mariette Driessens (VSOP, 荷兰), Michela Gabaldo (Fondazione Telethon, 意大利), Marlene Haffner (Haffner咨询, 美国), Virginie Hivert (EURORDIS – Rare Diseases Europe, 法国), Eric Hoffman (ReveraGen, 美国), Anneliene Jonker (IRDiRC, 法国), Sangeeta Jethwa (罗氏制药, 瑞士), Eri Matsuki (AMED, 日本), Ana Mingorance (Draecona咨询, 西班牙), Thomas Morel (Leuven大学, 比利时), Daniel O’Connor (英国药品和健康产品管理局), Anne Pariser (美国国立卫生院), Caridad Pontes (Autonoma de Barcelona大学, 西班牙), Ken Sakushima (日本医药品医疗器械综合机构), Maurizio Scarpa (MetabERN, 意大利), Richard Yang (ReflectionBio, 中国香港).
工作队采纳了110个可供孤儿药物研发项目使用的模块,并为每个模块制作了简明的概况介绍。介绍中包括了应用该项开发工具的关键信息,以及专家对其优缺点的评述。这些模块分为五个主要部分:监管信息约占总体数量的50%,包括申报路径、资格认定和政策激励;相关开发资源约占25%;最佳开发实践占15%;提前供药方案占6% ,即在药物上市之前为患者提供药品的方法;医疗技术评估机构为评估药物经济价值而制定的方法和程序占4%。

110个模块(资源和举措)分为 5 类,监管信息、开发资源、最佳开发实践、提前供药方案,以及医疗技术评价:
监管信息部分包含了大多数模块,约占50%,其中包括申报路径(Pathways)、资格认定(Designation)以及欧洲、日本和美国的孤儿药开发政策激励措施;
开发资源类别约占模块数量的25%,涵盖了各种类型的能够支持孤儿药开发的社会资源;
最佳开发实践占模块数量的15%,覆盖了罕见疾病领域已有的成功经验,从而在速度、质量或效率方面改善孤儿药物开发过程;
提前供药方案占6%,包括在当地监管机构批准药物上市前允许患者使用药物的途径。这些药物要么可报销,要么可免费提供;
医疗技术评价占4%,且主要由欧洲区域的政策文件构成(不包括仅限特定国家的工具),这部分内容涵盖了药物研发机构用于证明其药物社会经济价值的评估方法和监管程序。
在所有的模块中,监管文件大多是针对某个行政区域的,而开发资源和最佳开发实践多是全球通用的。ODDG工作组的专家对每个模块进行评估,从而衡量其与罕见疾病药物开发的相关性、可用性、适用范围,识别各个模块的利益相关者、推动者、价值输出,并给出专家提示、利弊分析以及最佳应用时间。这些资源均可在 www.irdirc.org 获取。
工作队成员对不同的模块进行了标注,确定了其在罕见疾病药物开发过程中的最佳用法,并将其与常见的药物开发概念整合,构成一个完整的药物开发框架(图3)。
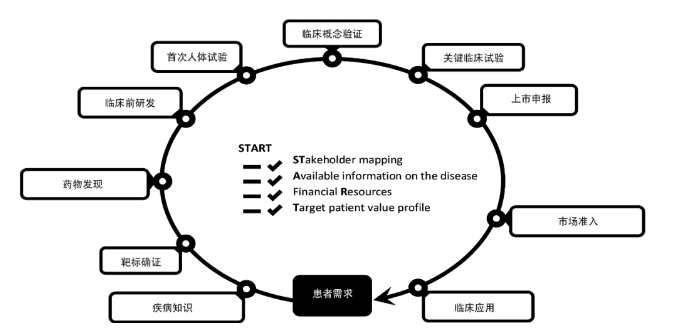
图3
为了确定如何最好地应用各个模块,工作组设定了三种典型情况:1)传统药物(即小分子或已知的生物制剂),在充分了解的罕见疾病中应用;2)前沿技术/药物在罕见病中的应用;3)针对尚未充分认识疾病的药物开发,其特征包括极低的患病率、散乱的疾病知识以及无法明确定义的终点或生物标志物(图4)。图5显示了这三种情况下不同模块使用情况的表示形式。

图4
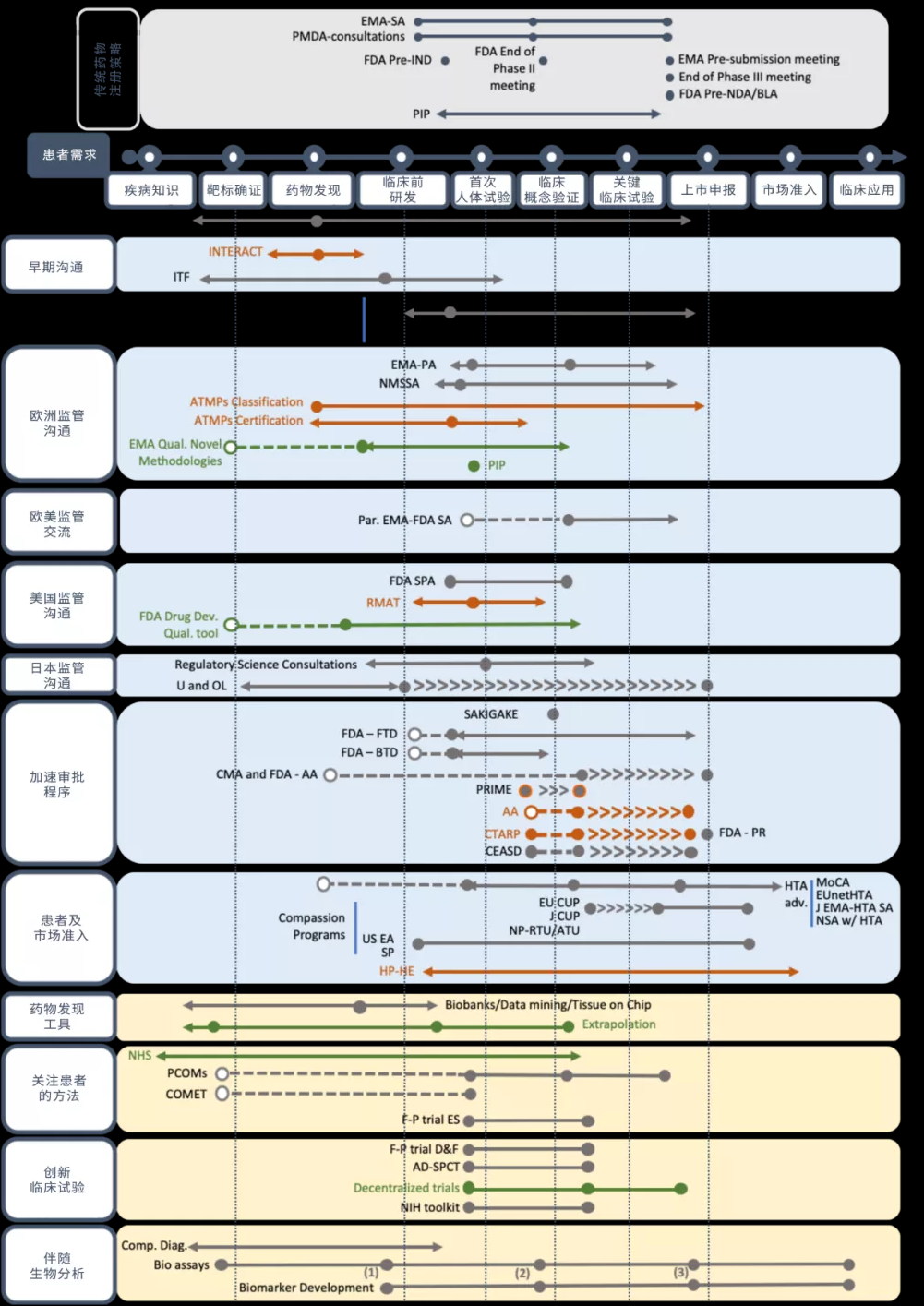
图5
如何启动(START)孤儿药物开发?
针对已经归纳整理出的所有模块,工作组也制作了另一份确认清单(checklist),以指导罕见病新药开发的启动。这项清单可为早期发现阶段(Discovery phase)的新药研发项目提供指导,也可为正在进行融资、合资以及许可交易的研发项目提供尽职调查方便的帮助。清单共分为五个部分,组成“启动”的英文单词"START":Stakeholder(利益攸关方)、Available information on the disease(有关疾病的可用信息)、financial Resource(财务资源)和Target patient value profile(目标患者价值概况)。
该清单的重点是帮助药物开发团队尽快建立疾病本身以及临床诊治方面的工作基础。使用差距分析(Gap analysis)方法,孤儿药开发团队可自行找到尚待完善的工作,例如未引入特定的利益相关者群体(如患者组织),或与缺少疾病相关的信息,如自然病史、基因型-表型相关性(genotype-phenotype correlation)、预测性生物标志物和临床终点。
及早加深在这些领域的认识,可以避免在药物开发的关键时期出现问题并导致药物开发的延误甚至失败。在关键临床试验(pivotal trial)的设计以及监管评估时,上述信息的充分掌握是至关重要的。在以上这些工作的基础上,根据患者的预期价值建立目标产品特征(Target product profile,TPP)即是启动阶段的核心任务。
监管和价值评估
孤儿药开发团队应尽早启动与监管系统的常规沟通,并尽量早于常规的强制性里程碑会议(如临床前、关键试验前、上市申报前会议)与监管机构进行沟通。这样做可以尽量前瞻性地优化药物开发过程,降低监管风险。
在许多情况下,监管战略的第一个里程碑是,只要能产生足够的支持证据,就立刻争取孤儿药物资格认定。在欧洲和美国已有成型的政策机制,协助孤儿药开发团队与监管机构进行沟通。例如 EMA Innovation Task Force和 FDA INTERACT 会议。在获得孤儿药资格认定之后,仍有很多监管流程和方案可以进一步提高药物开发团队与各监管机构的对话程度,其中包括EMA的protocol assistance、FDA的type C meetings以及与日本PMDA的咨询沟通。
在欧洲,向成员国监管机构寻求试验前科学咨询程序(pre-trial scientific advice procedures)所获得的益处经常被低估。孤儿药开发手册中的多个模块也涉及了前沿疗法的监管程序,包括欧洲的分类和认证程序、美国的 RMAT 资格以及日本再生医疗产品(regenerative medical products)的有条件、有时限的授权。
针对高度未满足医疗需求的产品,有多种审批加速程序来加快其上市,并且大多都适用于孤儿药开发。在提交针对性的申请前,应当充分理解这些加速审批程序所带来的程序收益以及资格认定所需要的材料,以便在早期临床阶段更全面地制定基于风险的上市前和上市后证据收集策略。
价值评估和报销策略可以在药物开发初期与欧洲医疗技术评估机构讨论。此类讨论可以是专门的会议,也可以是加入EMA 科学建议(EMA scientific advice)一起讨论。如果某个药物证明了其巨大的临床获益,孤儿药相关的资格认定往往可以让急需这些药品的患者更早用药——甚至在正式的监管和市场准入决定之前。这些机会来自于同情用药、指定患者用药计划(named-patient access scheme, 一种在欧盟框架下由医生负责,针对单个患者、不经监管部门批准的未上市药物用药途径)等多种特殊的监管途径。
药物开发资源和实践
工作组明确了多项可提高药物开发质量和降低药物开发风险的开发资源和实践,包括公开的药物发现工具、以患者为中心的实践、创新临床试验设计及执行方法以及生物标志物和伴随生物分析(companion bioanalytics)的开发。过往的IRDiRC工作成果总结了如何在小人群试验中获得有意义的数据[4,5],以及如何设计具有针对性且与病情更相关性的、符合临床试验及卫生经济学评估需求的终点或获益。在试验设计和终点选择等方面,患者及积极参与也是必不可少的。
结论
IRDiRC 的 ODDG 工作组汇总了供孤儿药物开发团队使用的工具,并制定了使用这些工具的策略。这些工作成果为药物开发团队提供路线图,尤其为不熟悉这一复杂过程的利益攸关人提供了重要的沟通工具。通过充分利用已有资源,可以大大避免药物开发过程的延迟,降低开发过程中的风险和成本,并提高患者和监管机构对孤儿药的进一步接受。
doi: 10.1038/d41573-020-00060-w
致谢
本文代表IRDiRC ODDG工作组和疗法科学委员会的集体智慧。除作者外,成员和撰稿人是A. Aiuti, D. Athanasiou, L. Conklin, S. Cheng, R. Conwit, M. Driessens, M. Haffner, E. Hoffman, S. Jethwa, E. Matsuki, A. Mingorance, T. Morel, A. Pariser, C. Pontes, M. Scarpa and R. Yang。我们还要感谢A.-L. Pham-Hung d’Alexandry d’Orengiani的支持。这项工作得到了欧洲合同"SUPPORT-IRDiRC"(N+ 305207)和"European Joint Programme on Rare Disease"(N+825575)的支持。
免责声明
本文中表达的观点是作者的个人观点,不可理解为代表其雇主/组织。
参考文献
1.Nguengang Wakap, S. et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur. J. Hum. Genet. 28, 165–173 (2020).
2.Boycott, K. M. & Ardigó, D. Addressing challenges in the diagnosis and treatment of rare genetic diseases. Nat. Rev. Drug Discov. 17, 151–152 (2018).
3.Austin, C. P. et al. Future of rare diseases research 2017–2027: An IRDiRC perspective. Clin. Transl. Sci. 11, 21–27 (2018).
4.Day, S. et al. Recommendations for the design of small population clinical trials. Orphanet J. Rare Dis. 13, 195 (2018).
5.Morel, T. et al. Measuring what matters to rare disease patients - reflections on the work by the IRDiRC taskforce on patient-centered outcome measures. Orphanet J. Rare Dis. 12, 171 (2017).
编译/Richard
编辑/张皓宇
病痛挑战基金会的官方媒体平台面向广大伙伴,长期征集有关罕见病领域的原创观察、评论类稿件,欢迎原创、翻译类文章投稿,邮箱 media@chinaicf.org 。
原文链接:https://www.nature.com/articles/d41573-020-00060-w
孤儿药物开发指南材料:https://irdirc.org/orphan-drug-development-guidebook-materials

- 遗传常识-纯合/杂合/半合子
- Dystrogen Therapeutics 研究性嵌合细胞疗法 DT-DEC01 治疗 Duchenne 肌营养不良症临床显示有显著的功能和生物标志物改进
- Sarepta 同时布局外显子跳跃、PPMO和DMD 基因治疗
- CureDuchenne 通过投资 Code BioTherapeutics 继续致力于下一代基因治疗
- 遗传性包涵体肌病的基因研究进展2019.05.025
- 糖皮质激素能延缓可行走DMD患者并发症发生
- 肌肉剪切波弹性成像可用于监测 DMD 进展
- 新报告显示 2021 年罕见病药物开发资金同比增长 28%
- 揭开药品价格的神秘面纱
- 跳跃基因的适应将CRISPR-Cas基因写入技术推向新高度

至爱微信服务号
全部评论