01疾病概况:
肢带型肌营养不良症(Limb Girdle Muscular Dystrophies,LGMD) 是一种高度异质的遗传性神经肌肉疾病。临床主要表现为骨盆带、大腿、肩胛带和手臂近端进行性无力,可能在任何年龄发病,早期会出现异常步态、跑步困难,随着时间的推移和疾病的进展,最终可能需要轮椅协助。这些患者血清肌酸激酶 (CK) 浓度通常升高,肌肉活检显示营养不良,例如肌纤维的退化或再生。虽然部分LGMD的临床表型可能重叠,但少数LGMD亚型具有心功能障碍、远端肌无力、小腿肥大、关节挛缩、不对称无力等特殊表现。
(图1 肩胛骨为与锁骨共同组成肩胛带)
LGMD是继Duchenne和Becker肌营养不良(DMD/BMD)、强直性肌营养不良和面肩肱型肌营养不良的第四大肌无力疾病,预估患病率高达1:20000,携带率高达1:150。最近的一项研究统计,LGMD2型约占84%,LGMD1型约占16%,其中LGMD2A占24.7%,LGMD2B占23.8%,LGMD2D占9.7%。图二统计了发表的文献研究队列中LGMD各亚型的占比情况。
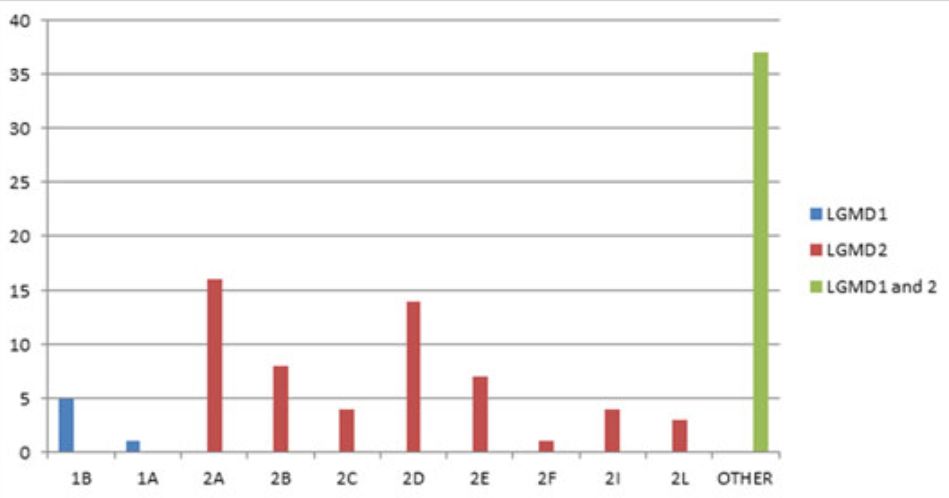
(图2 LGMD各亚型的占比情况)
02遗传学背景:
肢带型肌营养不良与基因突变引起的肌细胞蛋白功能缺陷有关,受累蛋白涵盖肌纤维所有结构,如细胞核、细胞质、肌小节、肌膜和细胞外基质。按照遗传方式,可以分为常染色体显性遗传和常染色体隐性遗传两种类型,每种类型根据致病基因又分为具体的亚型。LGMDs的类型通常用字母和数字来划分,这些字母和数字表明了被确认或怀疑的与这一型LGMD有关的基因,以及它的遗传方式。显性遗传的LGMD以LGMD1表示,约占10%。常染色体显性遗传的LGMD临床变异性较大,起病年龄较晚,进展相对缓慢,血清CK升高幅度相对较小。隐性遗传的LGMD以LGMD2表示,约占90%。和常染色体显性的LGMD相比,常染色体隐性的LGMD通常起病年龄更早,进展速度更快,血清CK相对较高。
03疾病分类:
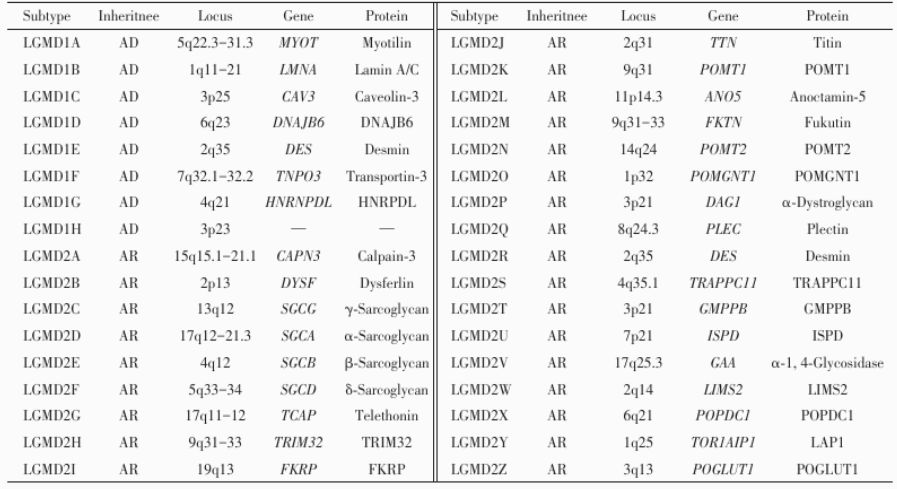
Classical subtypes of limb girdle muscular dystrophies and according gene/protein
| 经典分类 | 基因定位 | 基因产物 | 蛋白 | 相应疾病 |
|---|---|---|---|---|
| LGMD 1A | 5q31.2 | MYOT | myotilin | Z盘蛋白病 |
| LGMD 1B | 1q21.2 | LMNA | lamin A/C | 核膜蛋白病 |
| LGMD 1C | 3p25.3 | CAV3 | caveolin3 | 小凹蛋白病 |
| LGMD 1D | 7q36 | DNAJB6 | DNAJB6 | Z盘蛋白病 |
| LGMD 1E | 2q35 | DES | desmin | Z盘蛋白病 |
| LGMD 1F | 7q32 | TNPO3 | transportin 3 | 核膜蛋白病 |
| LGMD 1G | 4q21 | HNRNPDL | 异质核糖蛋白D样蛋白 | RNA结合蛋白 |
| LGMD 1H | 3p23p25.1 | 未知 | 未知 | 未知 |
| LGMD 1I | 15q15 | CAPN3 | calpain3 | calpain蛋白病 |
| LGMD 2A | 15q15 | CAPN3 | calpain3 | calpain蛋白病 |
| LGMD 2B | 2p13 | DYSF | dysferlin | 膜修复缺陷肌营养不良 |
| LGMD 2C | 13q12 | SGCG | γsarcoglycan | sarcoglycan蛋白病 |
| LGMD 2D | 17q21 | SGCA | αsarcoglycan | sarcoglycan蛋白病 |
| LGMD 2E | 4q12 | SGCB | βsarcoglycan | sarcoglycan蛋白病 |
| LGMD 2F | 5q33 | SGCD | δsarcoglycan | sarcoglycan蛋白病 |
| LGMD 2G | 17q12 | TCAP | telethonin | Z盘蛋白病 |
| LGMD 2H | 9q33 | TRIM32 | tripartite motif containing32 | 泛素化酶 |
| LGMD 2I | 19q13 | FKRP | fukutin相关蛋白 | αDG蛋白病 |
| LGMD 2J | 2q24 | TTN | titin | Z盘蛋白病 |
| LGMD 2K | 9q34 | POMT1 | proteinO甘露糖转移酶1 | αDG蛋白病 |
| LGMD 2L | 11p14 | ANO5 | anoctamin5 | 膜修复缺陷肌营养不良 |
| LGMD 2M | 9q31 | FKTN | fukutin | αDG蛋白病 |
| LGMD 2N | 14q24 | POMT2 | proteinO甘露糖转移酶2 | αDG蛋白病 |
| LGMD 2O | 1p32 | POMGnT1 | proteinOmannose β 1,2Nacetylglucosaminyltransferase | αDG蛋白病 |
| LGMD 2P | 3p21 | DAG1 | αDG | αDG蛋白病 |
| LGMD 2Q | 8q24 | PLEC1 | plectin 1 | Z盘蛋白病 |
| LGMD 2R | 2q35 | DES | desmin | Z盘蛋白病 |
| LGMD 2S | 4q35 | TRAPPC11 | transport protein particle complex 11 | αDG蛋白病 |
| LGMD 2T | 3p21 | GMPPB | GDPmannose pyrophosphorylase B | αDG蛋白病 |
| LGMD 2U | 7p21 | ISPD | isoprenoid synthase domaincontaining protein | αDG蛋白病 |
| LGMD 2V | 17q25 | GAA | α1,4glucosidase | 糖原累积病(Ⅱ) |
| LGMD 2W | 2q14 | LIMS2 | LIM and senescent cell antigenlike domains 2 | Z盘蛋白病 |
| LGMD 2X | 6q21 | POPDC1 | popeye domaincontaining protein 1 | 核膜蛋白病 |
| LGMD 2Y | 1q25.2 | TOR1AIP1 | torsinA interacting protein 1 | 核膜蛋白病 |
| LGMD 2Z | 3q13.33 | POGLUT1 | protein O葡萄糖基转移酶1 | αDG蛋白病 |
注:LGMD:肢带型肌营养不良
https://zfin.org/action/nomenclature/history/ZDB-GENE-061110-16
(图3 LGMD的分类)
1、肌聚糖病 Sarcoglycanopathies
包括α‐sarcoglycanopathy (LGMD2D), β-sarcoglycanopathy (LGMD2E), γ‐sarcoglycanopathy (LGMD2C), 和δ‐sarcoglycanopathy (LGMD2F) ,占AR遗传的儿童期LGMDs的68%,成人期LGMDs的10%。骨盆带肌早期受累,随后涉及到肩胛带,出现翼状肩,抬手臂困难。心脏受累程度各不相同,但都有心肌受累。在罗马和突尼斯人群中,LGMD2C亚型更常见。意大利10%的近端肌营养不良患者被诊断为肌聚糖病,其中52%的患者进展快速,48%的患者病情进展缓慢。
2、钙蛋白酶病 Calpainopathies
钙蛋白酶病calpainopathies由编码Calpain-3钙蛋白酶的CAPN3基因突变导致。Calpain-3蛋白与细胞骨架蛋白如titin和dysferlin相互作用,并在肌节重塑中发挥作用。Calpain-3功能丧失导致肌节异常并最终导致肌纤维死亡。患者临床表型可变,从轻微到严重。包括以骨盆带和后期肩带肌无力,脚尖走路为首发症状的Pelvifemoral LGMD (Leyden‐Möbius) 和以肩带肌和后期骨盆肌无力,血清CK升高为症状的scapulohumeral LGMD (Erb)。钙蛋白酶病calpainopathies占LGMD的15-40%。东欧,捷克共和国,西班牙,意大利,荷兰,英格兰北部和巴西等一些地区该亚型的患病率更高。
3、Dysferlin肌病
Dysferlin肌病是DYSF基因突变致使其编码的Dysferlin蛋白结构或功能异常而引起的。以骨盆和腰带肌肉早期无力和萎缩为特征。不涉及呼吸系统和心肌。起病年龄在11-40岁之间。Dysferlin蛋白在修复肌细胞膜,避免肌纤维变性的过程中起关键作用。Dysferlin肌病存在广泛的临床变异性。DYSF基因突变也可导致下肢远端后群发病的Miyoshi远端肌病、远端前群肌病、也有仅表现为运动不耐受或无症状高CK血症。各亚型的临床表现呈一种进展性状态,即从一种临床表现转为另一种临床表现。预估Dysferlin肌病占LGMD的5%-35%,在亚洲、中东、印度等地发病率高。
4、蛋白聚糖病 Dystroglycanopathies
蛋白聚糖病为一类由FKRP, GMPPB, ISPD, LARGE, POMGnT1, POMT1或POMT2基因突变引起的常染色体隐性肌营养不良症。这些突变的潜在致病机制是细胞外α-dystroglycan蛋白的糖基化水平降低。除了LARGE外其它基因的突变都与儿童或成人起病的常染色体隐性LGMD有关。此外,由DAG1错义突变导致的α-dystroglycan蛋白缺陷已被确定为与认知功能障碍相关LGMD的原因。几个基因突变引起的继发性糖基化异常也与先天性肌营养不良相关,包括Walker-Warburg综合征、Fukuyama型先天性肌营养不良和肌-眼-脑病。
5、核纤层蛋白病 Laminopathies
核纤层蛋白病是由编码核纤层蛋白A/C的LMNA基因突变引起的,包括常染色体显性的Emery-Dreifuss肌营养不良和伴心脏传导障碍的LGMD1B型等许多具有不同表型的病症。
6、小窝蛋白病 Caveolinopathies
小窝蛋白病由编码caveolin 3蛋白的CAV3基因突变导致,包括无症状的高CK血症,肌痛痉挛,波纹肌肉病rippling muscle disease和远端型肌病。这几种表型比经典的LGMD更常见。
LGMD病理学中涉及的几种重要病理机制包括抗肌萎缩蛋白-糖蛋白复合物的异常,肌小节,糖基化异常,囊泡和分子运输,信号转导途径和细胞核功能。这些通路之间不是完全分开的当然也不是完全重叠。事实上,一些疾病亚型是几个通路共同缺陷造成的。
04主要的辅助检查:
1、血清肌酸激酶 (creatine kinase, CK) 检测
LGMD患者通常可见血清CK水平升高,在肌聚糖病、Dysferlin肌病和小窝蛋白病中血清CK也存在异常升高。
2、肌电图和骨骼肌影像学检查
骨骼肌CT和MRI均为无创性检查方法,可以评价肌肉水肿、脂肪浸润、肌萎缩和肌肉肥大程度,以及判断肌肉受累模式,对诊断和分型具有指导意义。
3、肌肉组织活检
肢带型肌营养不良症肌肉病理改变主要呈现肌营养不良样病理改变特点,即肌纤维直径变异增大,出现肌纤维肥大、萎缩、坏死、再生和结缔组织增生,并伴有肌纤维分裂、镶边空泡、分叶状肌纤维等,部分亚型还可表现为肌病样病理改变。随着病程进展,肌纤维逐渐被脂肪和结缔组织替代。由于其发病机制涉及肌细胞蛋白缺陷,采用特异性抗体进行免疫组织化学染色或免疫印迹法测定蛋白质含量对特定亚型的诊断具有重要价值。
4、基因检测
通过基因检测发现致病性突变,最终实现分子生物学诊断是肢带型肌营养不良症的最终诊断和分型依据。目前二代测序技术检出率高达65%【12】,提示二代基因测序技术在肢带型肌营养不良的诊断中具有广阔的应用前景。
05实例解读
患者,男,21岁,双下肢无力8年余,腰椎弯曲,行走鸭步,Gower’s征,CK4147 U/L。肌电图检查上下肢NCV、F波未见明显异常。上下肢EMG肌源性损害。腰椎退行性变;腰1/2椎间盘变性。胸椎MR平扫未见异常表现。无家族史。肌营养不良待查。
全外显子组(WES)基因检测结果
该样本发现与其表型高度相关的基因突变。

突变细节:
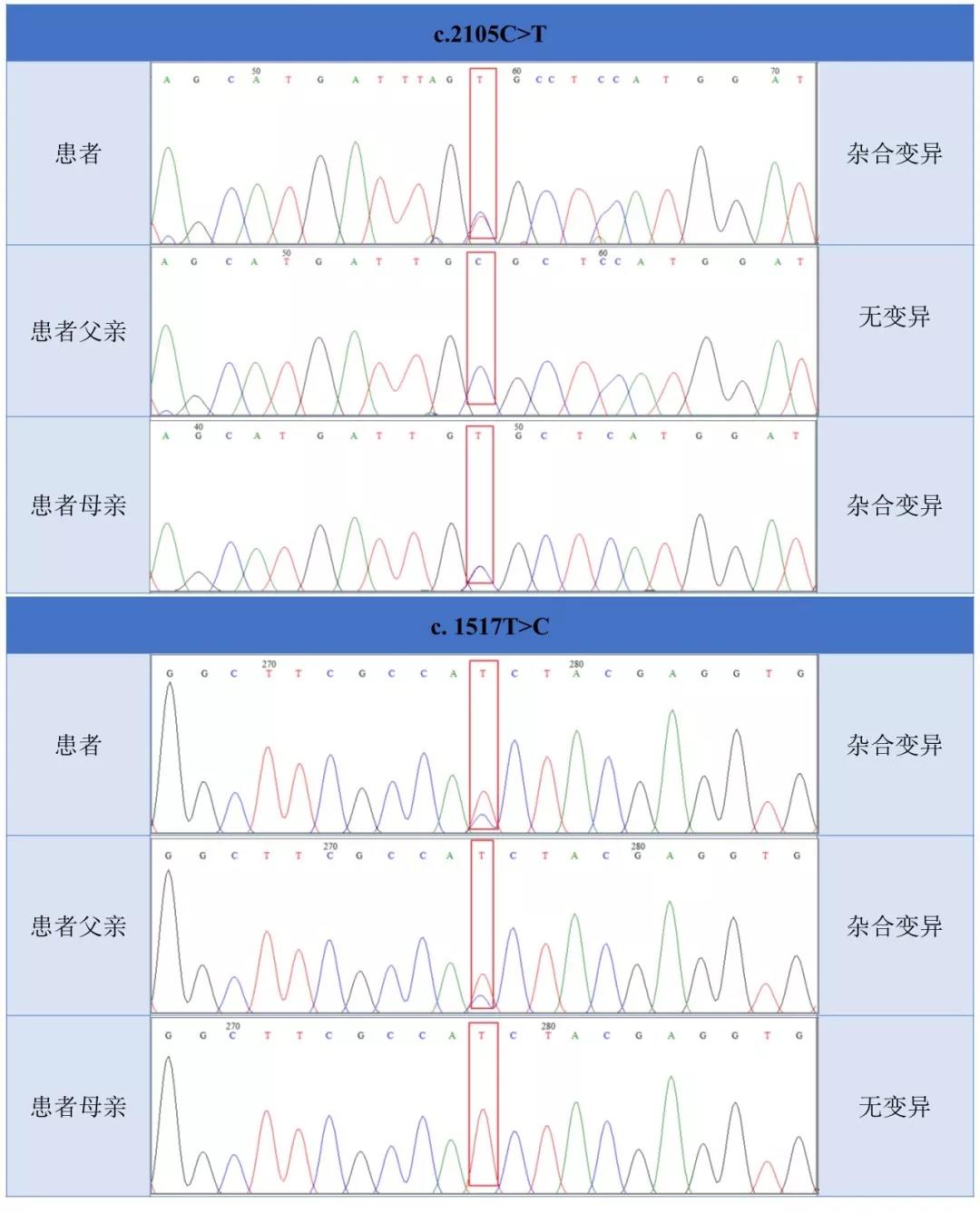
CAPN3, 外显子19/24, chr15:42702183, NM_000070.2:c.2105C>T (p.Ala702Val), 杂合子, 可能致病
- 此突变在gnomAD数据库、千人数据库、ExAC 数据库中正常人群未发现(或极低频位点)(PM2)。
- 该变异与Met792Ile 形成复合杂合,在患者中检出(PMID:17994539)。另外,在隐性遗传病中,在本例患者反式位置上检测到致病变异Ile506Thr(PM3)。
- 多种统计方法预测该变异会对基因或基因产物造成有害的影响(PP3)。
- 此突变被文献报道与肢带型肌营养不良2A型相关(PMID:27262448,9150160,17236769,16141003,19556129)。有可靠信誉来源的报告认为该变异为致病的, 但证据尚不足以支持进行实验室独立评估(PP5)。
CAPN3, 外显子11/24, chr15:42694001, NM_000070.2:c.1517T>C (p.Ile506Thr), 杂合子, 可能致病
- 此突变在gnomAD数据库、千人数据库、ExAC 数据库中正常人群未发现(或极低频位点)(PM2)。
- 在隐性遗传病中,在反式位置上检测到致病变异p.Ala27AlafsX7(PMID:25987458)(PM3)。
- 多种统计方法预测该变异会对基因或基因产物造成有害的影响(PP3)。
- 此突变被文献报道与肢带型肌营养不良2A型相关(PMID:25987458)。有可靠信誉来源的报告认为该变异为致病的, 但证据尚不足以支持进行实验室独立评估(PP5)。
Sanger验证结果
案例小结
LGMD2A型是肢带型肌营养不良中最常见的亚型之一,起病于3-30岁,儿童或成人早期起病者较为多见,多数患者表现为轻度至中度的进行性肢体无力,下肢重于上肢。临床上以鸭步和脊柱过度前凸为主要特征。髋、膝和肘关节可迅速出现挛缩,通常不累及面肌、心肌及呼吸肌群,少见腓肠肌假肥大。血清CK早期可增高5-20倍,以后可恢复正常。
CAPN3基因编码肌肉特异性钙激活蛋白水解酶calpain 3蛋白。该蛋白不属于结构蛋白,为胞浆蛋白,是肌纤维胞质内的蛋白水解酶,有一定的自溶性,在体外易于降解,所以常规免疫组织化学染色难以准确判断蛋白表达情况。尽管采用calpain 3/dysferlin联合免疫印迹方法对诊断具有临床价值,但是特异性和敏感性也有一定的局限性(60kDa条带缺失或减弱时,特异性约为94%,敏感性约为64%。30kDa条带缺失或减弱时特异性为88%,敏感性为58%(https://neuromuscular.wustl.edu),也有研究表明calpain 3蛋白可继发性减少),因此最终疾病的确诊需依赖基因检测。由于肢带型肌营养不良是一组遗传异质性和临床变异性极强的神经肌肉病,二代测序技术将在该疾病的诊断与鉴别诊断中发挥重要的作用。
06诊断流程:
因为其它一些同样以近端肌无力为特征的神经肌肉病比LGMD更为普遍,所以临床上应重点加以鉴别诊断。此类疾病包括Duchenne和Becker肌营养不良 (DMD/BMD),获得性肌肉疾病,如中毒性、内分泌和自身免疫性肌病,以及非肌肉疾病,如重症肌无力和脊髓性肌萎缩等。对于怀疑患有特定类型LGMD的患者,建议在进行肌肉活检之前进行基因检测,以确定最有可能解释临床表现的特定基因突变。例如,建议具有DMD/BMD临床表型但是DMD基因检测阴性的患者都要进行LGMD2I的基因检测,具有Emery-Dreifuss肌营养不良表型的患者进行LMNA基因检测。
LGMD的表型复杂使得建立有效的临床诊断流程至关重要。按照肌无力的类型、遗传方式以及临床和肌肉活检特征可以有效地将鉴别诊断范围缩小到具体的疾病。整体的流程见图5,详细细节请参照图6-9。
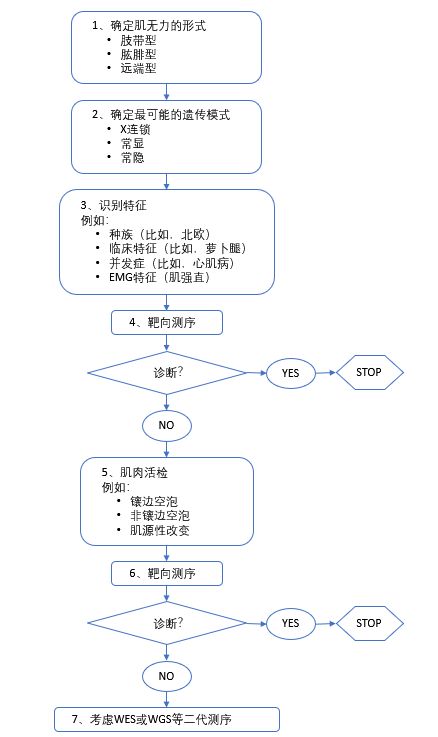
(图5 LGMD的临床诊断流程)
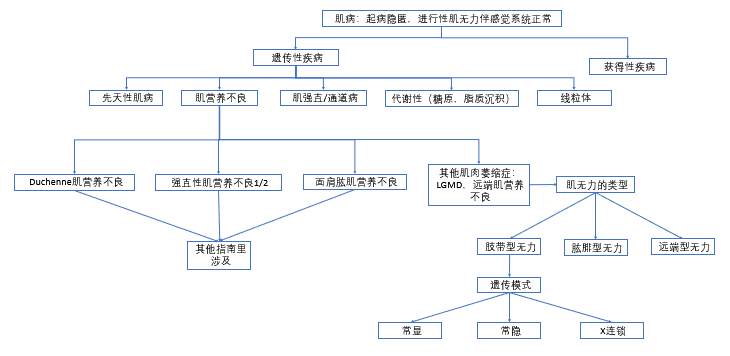
(图6 肌病的经典分类)
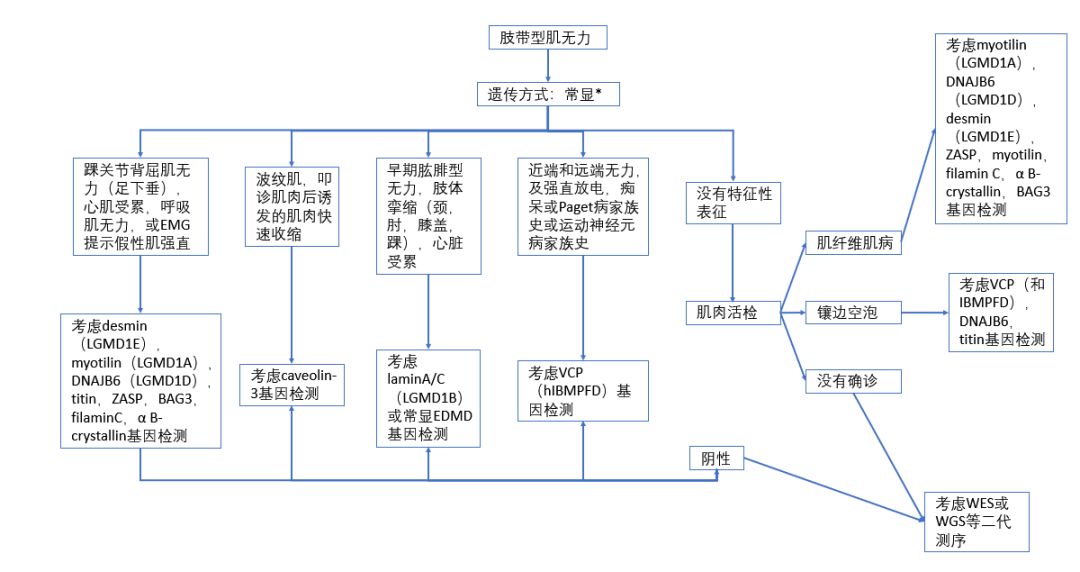
(图7 怀疑常染色体显性遗传患者的诊断流程)
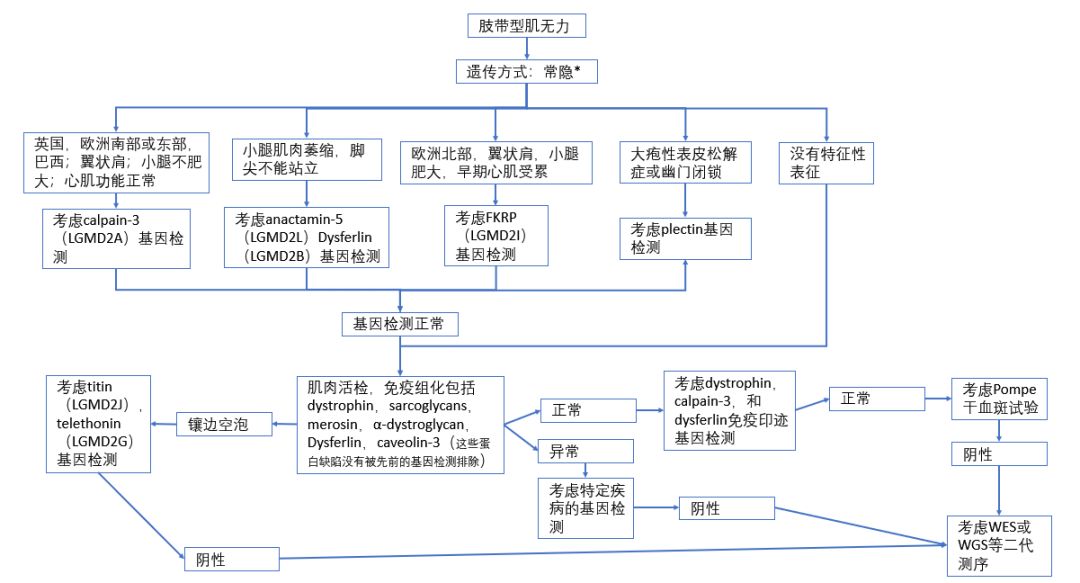
(图 8 怀疑常染色体隐性遗传患者的诊断流程)
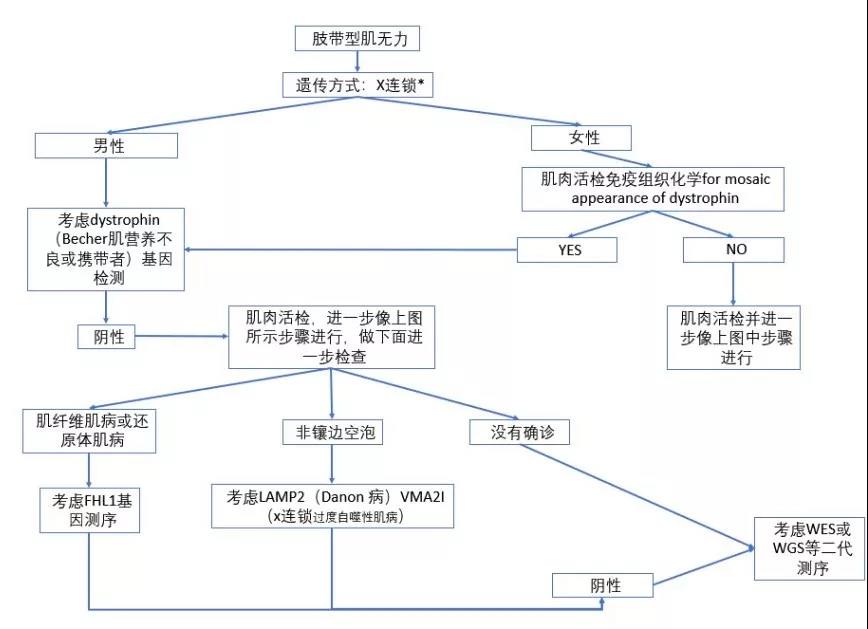
(图9 怀疑X连锁患者的诊断流程)
07 治疗、管理与预后
目前针对LGMD没有有效的治疗方案,一般为支持性治疗,目标包括保持活动能力,管理相关的并发症,最大限度地提高生活质量。
康复治疗和运动:挛缩可能导致严重残疾,所以LGMD管理的重要方面是预防挛缩,应尽早建立被动拉伸物理治疗方案。
心脏并发症:LGMD的许多亚型具有相关的心脏受累,包括LGMD1A, LGMD1B, LGMD1D, LGMD1E, LGMD2C至2K和LGMD2M至2P。一般情况下,患有这些亚型的患者可能没有明显的心脏病症状,但是仍存在心血管疾病或猝死的风险。因此,患有以上亚型的LGMD患者以及没有明确诊断的LGMD患者应该进行基础的心血管疾病评估,包括心电图、超声心动图或心脏MRI等。患有LGMD2C, 2D, 2E和2F (sarcoglycanopathies) 和LGMD2I且心脏功能正常的患者应每两年进行一次心脏随访;心脏检查结果异常的患者应每年进行一次随访。LGMD1B患者应该进行类似的随访,如果无症状则每两年增加一次动态心电图监测,如果心脏检查异常则每年增加一次。所有患有合并心脏受累的LGMD患者应转诊到心脏病专科。心脏移植手术已经在LGMD1B患者和其他合并严重充血性心力衰竭患者中取得成功。
肺部并发症:一些LGMD(例如LGMD2I)与呼吸或口咽肌无力有关,随疾病进展,呼吸衰竭风险增加。有肺部并发症风险的患者在诊断时应进行肺功能检查或转诊进行肺功能评估。
吞咽和喂养困难并发症:与LGMD相关的吞咽困难和手臂无力可能导致患者无法获得足够的营养。摄入不足、吞咽问题或体重减轻的患者应通过吞咽研究进行评估或向胃肠病学家咨询。通过改变食物稠度或放置饲管,增加营养摄入。
骨科并发症:患有LGMD的个体可能会有更高的脊柱畸形的风险,包括脊柱后凸或脊柱侧凸。如果需要,发生脊柱畸形的患者应转诊到骨科专家进行评估和手术,以保持最佳姿势,活动能力和心肺功能。
临床试验:目前以基因导入、外显子跳跃、成肌细胞移植、生长激素等策略治疗各种LGMD亚型的研究正在开展中。然而,这些干预措施的有效性和安全性仍有待确定。
LGMD的预后多样。大多数常染色体隐性遗传的LGMD患者发病早,最初出现肌无力症状,在儿童期出现残疾。常染色体显性遗传的LGMD患者,肌无力可能要到成年后甚至更晚才会显现出来。除了少数几种亚型表现出进展快速或不对称肌无力,其余通常均进展缓慢,且表现为对称性肌无力。
通过这些我们了解到LGMD是一组较为常见的遗传性神经肌肉疾病,其临床表型具有高度变异性,存在30多种亚型,这些给此类疾病的诊断带来挑战。